Introduction:
To date, therapeutic advances in treating central nervous system (CNS) disorders have been incremental at best. As the global population increases and older demographics begin to make up a disproportionately larger percentage, CNS diseases like neurodegenerative and pain disorders, are poised to increase in prevalence. More than 25 million people today are living with Alzheimer’s Disease (AD) or related dementia, and estimates suggest that this number will double every 20 years.1 It is likely inevitable that as global life expectancy continues to increase, so will the likelihood that more individuals will face some sort of chronic neurological issue.
Current practices for preclinical drug development in neurological disorders typically require extensive animal modeling. In most countries, this is a legal requirement before entering human trials. However, these experiments rarely provide adequate translational power, particularly in disease modeling and efficacy studies, and are a large factor in the lack of FDA-approved therapies for treating chronic neurological diseases. Unproductive use of animal testing both increases cost and extends the timeline of reaching the clinic, while also wasting animal life and endangering trial subjects, as discussed in our previous white paper.2 Here, we will focus on the challenges of modeling neurological diseases and explore the role of induced pluripotent stem cell (iPSC) technology to improve the translational capacity of drug discovery in neurological diseases, particularly for chronic and heritable conditions.
II. Understanding neurological disease modeling
The term neurological disorder spans a wide variety of conditions, as it includes all disorders characterized by malfunction of the central and/or peripheral nervous systems. Despite huge differences in the etiology of neurological diseases, the majority of these disorders share one common trait: the vulnerability of specific neural or glial cells. This vulnerability can manifest in symptoms such as seizures, muscle weakness, cognitive decline, and partial to complete paralysis. Even though diverse neurons and cell types are affected in the various disorders, it is generally accepted that common pathological events lead to degeneration and cell death such as protein misfolding, organelle dysfunction, and chronic upregulation of inflammatory signaling.2 Several studies have evidenced which pathways and genes are common between various disorders allowing for more generalized treatment.3-5 A more in-depth understanding of the individuality of neurological disorders may provide crucial information on specific diversity in responses, such as why a broad treatment i.e. cell replacement therapy, may not be equally effective for diverse neurological disorders. The difficulties of researching the human nervous system and the intricate interplay between disease causation, pathology, and phenotype hinder the quest for treatments and cures for diverse neurological disorders.
There is little debate that biological aging is one of the most common and important risk factors in most neurodegenerative diseases. Aging is macroscopically correlated with gray and white matter atrophy, and ventricular enlargement6, and histologically correlated with reduced dendritic connections and neuronal death.7 While the pathological process of atrophy has been hypothesized to be mediated by aggregation of toxic proteins such as extracellular amyloid-β (aβ) and intracellular phosphorylated tau (pTau), as observed in AD, these protein aggregates also accumulate in clinically healthy older individuals8–11, in the absence of behaviors associated with any aforementioned degenerative conditions. Similarly, the decline in human brain function throughout aging is similar to that of other organs, implying that molecular underpinnings upstream of cell death and senescence are influenced by pan-cellular mechanisms of aging rather than tissue-specific protein dysfunction.12 This has been in focus recently with the hopes of Yamanaka factor-based therapies as a tool to combat aging and several chronic diseases.13 Thus, understanding the basic mechanisms of aging in the CNS may be a constructive way to approach studying the pathology of chronic CNS diseases. It is arguable that iPSCs, which are derived by manipulating the expression of Yamanaka factors, have the potential to model these chronic processes better than animal models.
iPSC models to study neurological disorders
For decades, a culture of primary rodent neurons was the predominant cellular model system for mammalian neurobiology. In the last few years though, we have seen a widespread increase in the use of human neuronal systems. These efforts stem from the long-held promise that iPSCs14–17 will deliver large and reproducible quantities of relevant human neural cells suitable to support the development of new therapies. Human datasets that support the scientific rationale for new targets have become a prerequisite for attracting the attention of biopharmaceutical companies and early-stage investors, and relevant in vitro models are required to assess the therapeutic properties of candidate drugs in a human cellular context. Precision medicine initiatives and changes in the drug regulatory landscape are providing additional impetus to develop solutions for use of high-quality, patient-derived cells in a reproducible and controlled environment18.
A strong argument can be made that the major hurdle in the development of treatments for neurodegenerative disorders is the current reliance upon animal models. Due to the nature of chronic diseases, where the underlying anatomical dysfunction often precedes behavior, it makes it difficult to elucidate biological targets without the tracking of underlying damage and knowledge of how it correlates with specific behavioral deficits. While small-animal models allow for investigations of disease progression and assessments of complex behavior that are unattainable in vitro, the human-specific nature of many diseases greatly impedes translatability - a catch-22. Recent advances in humanization and recombinant challenge viruses have increased the number and sophistication of available models19. Still, in vitro studies often suffer from the difficulty of modeling CNS cell types. Due to the paucity of live brain biopsy tissue, primary culture of human CNS cells is challenging at best and completely infeasible at the scale necessary to perform drug screens or examine synaptic-glial interactions. In addition, immortalized neural-like cell lines fail to replicate primary functions of the cells they model. While monocultures are useful for determining cell-specific effects of a given treatment, they also fail to recapitulate the non-cell autonomous nature of several disease states, as is the case in most disease or organ-specific models.
3D Modeling
Compared to 2-dimensional (2D) monolayer cell culture, 3D culture systems more accurately recapitulate CNS in vivo conditions from a transcriptional, anatomical, and electrophysiological perspective.20–22 Brain organoids, which are self-organizing neural structures that can mimic human fetal brain development, have now been used to develop alternative models of Alzheimer's disease, Parkinson's disease, motor neuron disease, and frontotemporal dementia because they recapitulate important neuropathological hallmarks found in these disorders.17 Despite these early successes, various limitations in brain organoid models must improve to better match human tissue features, such as relative tissue immaturity, lack of vascularization, and inadequate cellular diversity present in this culture system. As these obstacles are overcome, brain organoid models, enhanced by traditional and emerging molecular and analytic methods, will likely be able to uncover the pathophysiological causes of neurodegeneration and develop novel treatments for a variety of illnesses.
Organoids, by their very nature, appear to be an appealing system for understanding the interplay between these numerous pathways that contribute to neurodegeneration in light of this knowledge. The ability to construct youthful brain tissue with a lifetime of genetic events is enabled by reprogramming adult cells to form iPSCs. The likes of Sergiu Pasca and several other have assessed forebrain organoids that are >600 days old,19 roughly equalling the lifetime of a wild-type typical mouse used in studying behavior and aging. This creates a temporally condensed system in which pathogenic molecular and cellular mechanisms can be investigated upstream of protein aggregation and cell death, and in which the entire pathological cascades can be scrutinized and characterized at every step by -omics technologies to establish causal relationships between genetic/sporadic changes and neuronal health (Fig. 1).23
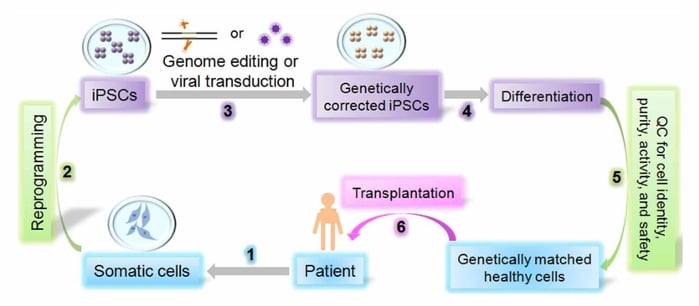
Figure 1. Illustration of iPSC production cycle including somatic cell reprogramming, quality control, and patient specific treatment.23
iPSC-derived cerebral organoids from older patients with “sporadic” neurodegeneration provide an attractive avenue to study these relationships, which is something not possible in static pathological tissue from human brain or via inference of pathological findings from animal neurodegenerative models. These organoids developed both extracellular β-amyloid plaques and neuronal aggregates of phosphorylated tau - findings that are inconsistently observed in two-dimensional neuronal cultures or animal models6. This is a timely innovation, as multiple potential drugs for Alzheimer’s disease developed in animal models have failed late-phase clinical trials.24–26 Biogen’s recent struggles with Aduhelm further highlight the need for more definitive proof-of-concept demonstration in efficacy, before launch. This 3D culture method could be used to evaluate and nominate new medicinal compounds as a complementary and scalable approach.
Why are iPSCs positioned to be uniquely successful for studying neurological disease?
The best iPSC-based models and screens take advantage of unique properties of the differentiated cell type. Vincent and colleagues laid out the phenotypic screening "rule of 3" as a guiding premise for evaluating the translational value of phenotypic screens27,28. They proposed that the best phenotypic screens show a combination of (1) high disease relevance of the assay system; (2) a disease relevant stimulus to produce the expected phenotype; and (3) an assay readout that correlates with the clinical endpoint. This does not imply that iPSC-based models replicate in vivo-biology with high fidelity, a goal that is seemingly impossible in current times. For usefulness, they need only capture the complexity of the in vivo phenotype being studied (in other words, the assay is “fit for purpose”). Herein lies a critical set of problems: we neither have a relevant disease model and disease-relevant stimulus, nor an appropriate clinically-relevant readout as it pertains to most neurological disorders. Direct measurement of disease-relevant proteins or functionally important changes in electrical properties of neurons are often the most relevant assay endpoints. Weak, low-penetrant, or environmentally induced phenotypes may be difficult to measure robustly, and some phenotypes associated with late-onset disorders may not be revealed in the absence of conditions that induce cellular stress or aging. Representing human disease relevance is a critical first step in creating a translational system that links the mechanisms underlying the assay phenotype directly to the same molecular drivers found in the selected preclinical disease model and ultimately to the clinical features to be measured in patients.17 The strength of the system of translatability defines the likelihood that a therapeutic drug discovery effort will yield meaningful results.29
The strongest case for using an iPSC-based model is its presumed physiological relevance. The fundamental advantage of iPSC-derived cell types is that they can be human or the closest model that exists to human - a point that cannot be overstated as it may be the single strongest argument for mass adoption. This is particularly important in the field of neuroscience, in which increasing numbers of examples demonstrate that human neurons rely on species-specific genes and mechanisms.30,31 Human primary neurons are not as widely available, and rarely in sufficient quantities to support screening for therapeutic drug discovery. Differentiated human iPSCs routinely achieve gene expression signatures that squarely align the differentiated cells with many appropriate in vivo target brain cell types, although they fail to achieve all the appropriate hallmarks of a comparable, fully mature brain cell32. Despite their immature phenotype, iPSC-derived neurons are capable of fundamental neuronal functions, including firing of action potentials and release of neurotransmitters as well as interaction with other CNS cell types in vitro.33 While the field will continue to strive for cell types that better match those found in vivo by incorporating an aging component, an iPSC-derived neuronal system remains a better model of a terminally differentiated and native-human neuron than immortalized or engineered cell lines. The ability to differentiate cells carrying specific mutations which are derived from a patient or that are CRISPR engineered makes it possible to directly evaluate the impact of disease-relevant mutations. These are the features that can make developing an iPSC-based screen worth the added effort and expense.
III. iPSC Challenges
Modeling
The use of human induced pluripotent stem cells (hiPSCs) to simulate specific CNS cell types and neuronal subtypes has advanced significantly over the last decade.34 These human cells are differentiated into a state that is morphologically, transcriptionally, and functionally identical to their main counterparts, including neurons, astrocytes, microglia, macrophages, and endothelial cells, using transcription factors or small chemicals. Adaptations of these individual cells in a system whereby these key players in the CNS can interact with each other are becoming more prevalent, with several multi-cell type CNS in vitro systems now available.15 While these systems are a first step in the generation of multicellular CNS cultures, standard practices currently do not assess systems where there is biological exchange across a blood-brain barrier (BBB), which is a critical part of the drug discovery process for CNS therapeutics. The BBB regulates the delivery of oxygen and nutrients to the brain, but more importantly selectively regulates active and passive transport of circulating substances in the blood. Thus, it provides a barrier for protecting the central nervous system from potentially harmful toxins or infections. However, in the context of neurological drug development, the BBB is an obstacle that can hinder clinical success. Therefore, development of 3D models that can incorporate the macroscopic function of the BBB is important to determine drug viability for treatment of neurological diseases. 3D models that incorporate brain macrostructures and replicate the BBB will confer several advantages for in vivo modeling and allow for examination of the effects of peripherally administered substances to penetrate the CNS. To this end, integrating microglial-like cells and other CNS immune cells into existing 3D and multicellular CNS organoid models offers an opportunity to observe infected human microglia in a spatial context with neurons, other glia, and larger brain structures.35
Even the most enthusiastic supporters of iPSCs admit that cultivation and differentiation of these cells can be difficult. They're also more expensive and time-consuming to use than primary rat neurons or immortalized cell lines. In theory, iPSC-derived cells can be employed in screens involving any type of modality, including small molecules, antibodies, or nucleic acids (anti-sense oligonucleotides, shRNAi, CRISPR). The design and validation of an iPSC-based model and/or screen should be carefully considered to ensure that the increased expense and labor are justified by the projected benefits gained by using the cells. Though given the synergistic potential of iPSC systems with current advances in sequencing technology, they may provide the capacity to rapidly advance how we model neurological disease. Furthermore, the nature of these technologies, especially with respect to sequencing and automated manufacturing protocols, will capitalize on the predictive decrease in cost as production scales (Moore’s law) and this could pave the way to iPSCs as the de facto in vitro system for preclinical studies.
While differentiating and maintaining iPSC models can be expensive and time consuming, these challenges can be addressed by increasing differentiation efficiency, allowing for more rapid differentiation and high-throughput iPSC modeling, which is necessary and, excitingly, sufficient to delineate the complex cross talk between all the diverse cell types in the CNS in different disease states. Obstacles for the usage of iPSC-derived models are rapidly eroding. The availability of patient-derived iPSC lines resulting from US and European initiatives has made tools from government-sponsored research available to more scientists and contributed to widespread use of iPSCs.36 The advent of using CRISPR and other gene editing tools to engineer iPSCs enables researchers to make paired patient mutation lines and isogenic control lines.27 Additionally, CRISPR engineering has enabled generation of fluorescently-tagged iPSC lines that define specific subcellular compartments, or with reporters that indicate the activation of specific genes or pathways (e.g. Allen Cell Collection).37,38 Single-cell nucleic acid sequencing (scRNA-seq) combined with lineage tracing and downstream computational analysis is rapidly expanding our understanding of developmental biology and allowing the generation of more specific and efficient iPSC-differentiation protocols.39,40 Together, these advances in biology and software have contributed to an increase in the use of iPSC-derived neurons as an alternative to transformed cell lines and primary rodent neurons in the basic research community.
Scalability
A big part of cell and gene therapy production involves moving components of production cell lines (e.g. cells, media, small molecules, or beads) around a process. This typically requires human intervention with a high level of expertise, and this approach is both slower and less capital efficient than developing automated systems that optimize speed of workflow, operate continuously, and move through the steps with limited human oversight. However, this is perhaps an oversimplification of a possible solution to a complex manufacturing problem. It is something that is being addressed by a growing ecosystem of iPSC-focused biotech companies, which we will explore further in a future write-up. Currently, a drug developer might spend four years and $150 million USD to set up 150,000 square feet of sterile manufacturing. Companies like Healios and Megakaryon who are manufacturing products using iPSCs, may end up requiring far more infrastructure to build out a facility that meets the scalability of their product’s demands than originally thought. Developing standardized cGMP-compliant manufacturing protocols that can maximize for scalability and cost per unit, is paramount. This not only benefits the biopharmaceutical enterprises directly involved with this specific aim, but also has the potential to drastically change the acquisition cost of cell lines for research and enable a path to democratization of iPSC technology as an accessible cell-culture tool for research centers and laboratories globally.
Quality Control
Generation and scaled-manufacturing of iPSC-based cell therapies is complicated and presents a barrier for clinical use of autologous or allogeneic iPSCs. However, some of these challenges may be more pronounced for autologous applications because the cell quality testing is more rigorous and therefore does not allow for as large of batch production. This will also contribute to increased costs with this approach.41 One of the challenges associated with iPSC-based therapies is the process of generating iPSCs from somatic cells. The time and labor required for cellular reprogramming and cyrobanking are highly dependent on the method used for cellular (Fig. 1).41
Desirable reprogramming methods that result in high efficiency with various somatic cells types, like DNA-targeting methods (e.g. Adeno-associated virus, Lentivirus) work effectively in the CNS. However, there is often a large time requirement to serially sub-culture cells until vector clearance is achieved. mRNA-based reprogramming, on the other hand, is regarded as a zero-footprint approach since the exogenous mRNA sequences that are transfected to the cells have a short half-life and cannot integrate into the transfected cells' genome. While still in early adaptation, this method may be one that optimizes for speed and time, especially as payload efficiency increases with better delivery methods. Regardless of the strategies employed to generate iPSCs, a standard of acceptable quality criteria must be clearly defined (Fig. 2). Given the degree of quality control steps involved in iPSC generation, it may be difficult for different regulatory agencies to establish consistent guidelines.
Autologous vs. Allogeneic Cell Generation
Reprogramming technology supports the development of both patient-specific, autologous and donor-derived, allogeneic iPSC-based cell therapies. For autologous iPSC therapies, a patient’s somatic cells are harvested from a skin biopsy or blood sample, and then reprogrammed and differentiated into a therapeutic cell type. Conversely, an allogeneic iPSC-based therapy relies on starting from one donor to generate a large number of iPSCs, making one larger batch that can be banked and then used in the treatment of many patient doses. The production process can therefore be scaled and ultimately reduce the cost of each dose. In both of these instances, the manufacturing process is conceptually similar and comprised of somatic cell acquisition, reprogramming to achieve pluripotency, expansion of iPSC line, and collection of cells to generate a bank (Fig. 1). The banked cells can then be used for directed differentiation to a therapeutically-relevant cell type. In this regard, the implementation of appropriate assays for in-process and final release testing is critical in order to meet cGMP compliance and track the critical quality attributes of the product as it’s being manufactured and ready for release (Fig. 2).41
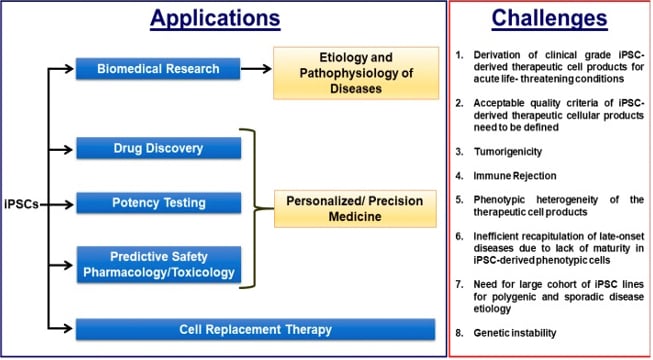
Fig. 2 – Applications for iPSCs and the challenges to executing these applications.41
A challenge for iPSC-based therapies is the efficiency and robustness of directed differentiation processes. These differentiations are typically developed based on a small number of clonal iPSC lines. To build a reproducible, reliable, and safe manufacturing process for autologous iPSC therapy, the differentiation process will need to be re-optimized and a definable standard of quality criteria needs to be set (Fig. 2). Though autologous cell therapies may serve certain patient populations better in many ways than allogeneic strategies (e.g. reducing odds of immune rejection, conferring better cellular integration, etc), it will also increase the time and cost of delivering this therapy to the patient, perhaps making it prohibitively expensive. Though, arguably, this may be necessary when considering applications for chronic CNS diseases, especially those of sporadic etiology where patient specificity is of high importance. Accomplishing this will also require a large cohort of iPSC lines to study such disease, further adding to the cost and time burden (Fig. 2).
Unlike autologous cell therapy, allogeneic iPSC-based therapies can provide an inexpensive, “off-the-shelf” solution, where there is a consistent, quality-assured product. This can be more appropriate for instances where peripheral administration of immune progenitor cells can facilitate CNS immune cell replacement, as has been observed in hematopoietic stem cell transplantation for microglial replacement.42 Another consideration for the development of allogeneic iPSC-based therapies is that for disease indications where the administration site is not immune-privileged, immunosuppressants cannot be administered, an HLA-matched or HLA-knockout iPSC could reduce the transplantation risks/complications.
Approaches for reduction of the cost of iPSC-based treatments
Despite cost averaging for large drug product batches, the cost of iPSC-based therapies could be prohibitive due to a number of factors, including single vendor-supplied specialty reagents, the need for highly skilled and time-intensive technicians throughout the process, suite utilization time associated with a long manufacturing process, and batch failure due to variable efficiencies from non-robust differentiation protocols. The transition from 2D cell culture systems to space-efficient, closed 3D computer-controlled suspension cultures will help incorporate automated methods and improved control of critical process parameters and allow for more consistent production of iPSCs and their derivatives without as much need for human intervention. Not only would this allow for increase the fidelity of representative 3D CNS these systems, but these cell cultures will have longer life cycles and enable protracted studies of disease under normal conditions.19 Along with transitioning cultures to 3D suspension-based processes, it is important to develop an optimized and robust process that is cost effective and that can perform reproducibly in manufacturing.
Emerging iPSC-derived technologies
The differentiation ability of iPSCs has been one of the primary drivers in the cell and gene therapy field. However, there are a number of emerging areas that may expand on the seemingly unlimited potential of iPSCs. These areas include: 1) organoid-generation for development and drug screening 2) iPSC-based immunotherapeutics and vaccines, and 3) iPSC-derived exosomes. Firstly, based on the observation that iPSCs and neural cells share a number of cellular and molecular properties, it is reported that inactivated iPSCs can elicit an immune response through the expression of similar surface antigens. Additionally, it was observed that the effect of this ‘iPSC-derived vaccine’ could be transferred from one ‘inoculated’ mouse to a diseased mouse, with sustained anti-tumor effects.43 Finally, the derivation of donor-matched organoids, from a single iPSC line, can have large implications for understanding drug interactions/metabolism with different patient populations. This can result in optimally titrated drug doses, as well as enable personalized medical interventions, a feature that will be critical to drug development of certain CNS diseases.
Excitingly, as the iPSC field continues to evolve and newer, innovative strategies are introduced, the fidelity with which the human CNS and other human organ systems are modeled will continue to asymptotically approach in vivo conditions.
IV. Conclusion
Although the discovery of iPSCs has already had a significant impact on the area of cell and gene therapy, their full potential has yet to be realized. The creation of universal, non-immunogeneic iPSC banks, as well as the utilization of scalable, automated, closed cell culture systems, would support the standardization of large-scale, manufacturing platforms for the cost-effective synthesis of large batches of therapeutic cells. Additionally, emerging iPSC-based technologies support both non-clinical and clinical use cases beyond the current focus on cell-replacement therapies. There is momentum already working in favor of iPSCs, particularly in adjacent fields that facilitate iPSC platform development, such as blood banking, transcriptomic and proteomic sequencing, where increases in both scale and affordability are being realized. Additionally, more and more individuals are starting to bank amniotic or cord blood, which can act as highly amenable substrates for iPSC synthesis and further decrease the cost compared to iPSC generation from further differentiated cells like skin fibroblasts.44 These developments will also help progress the therapeutic discovery of neurological diseases, particularly chronic degenerative diseases that will likely require long-term biomarker tracking and would benefit from using initial cell populations that are less likely to have age-prone genetic mutations.
Applying these novel modeling strategies in combination with improved gene editing technology and increased data sharing efforts, shows promise in unlocking disease modifying therapies for neurological diseases. Indeed, gene editing platforms have become more precise, and some are currently under clinical testing for treatment of certain diseases with known genetic targets. However, there are still gaps in our knowledge in how to apply gene therapies to certain neurological diseases. Efforts to characterize and share comprehensive public resources of tissue-specific gene expression like the Genotype-Tissue Expression Consortium45 funded by the National Institute of Health (NIH) aim to establish a database to study genetic variation, expression, and molecular phenotypes of multiple reference tissues, including multiple regions of the brain. These sorts of data sharing efforts become critical references when creating new iPSCs paradigms to model the CNS. This will also help standardize data sharing to foster best standard practices can improve generation, characterization, and modeling of the CNS.
Among the sea of considerations for improving CNS drug development and iPSC disease modeling that would be impossible to capture in one article, we touched on several present challenges and strategies that address these obstacles. iPSCs present a substantial opportunity to increase the translational power of preclinical/translational models and improve neurological disorder drug discovery efforts. Not only could this increase our fundamental understanding of CNS biology, but it can increase the odds of breakthroughs in a therapeutic area in which the industry has struggled to address a considerable unmet need.
References:
1 Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111-128.
2 Chi H, Chang HY, Sang TK. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int J Mol Sci. 2018;19(10):3082. doi:10.3390/ijms19103082
3 Calabrese G, Molzahn C, Mayor T. Protein interaction networks in neurodegenerative diseases: from physiological function to aggregation. J Biol Chem. Published online May 24, 2022:102062. doi:10.1016/j.jbc.2022.102062
4 Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement Transl Res Clin Interv. 2018;4:575-590. doi:10.1016/j.trci.2018.06.014
5 Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43-51.
6 Grenier K, Kao J, Diamandis P. Three-dimensional modeling of human neurodegeneration: brain organoids coming of age. Mol Psychiatry. 2020;25(2):254-274. doi:10.1038/s41380-019-0500-7
7 Dumitriu D, Hao J, Hara Y, et al. Selective Changes in Thin Spine Density and Morphology in Monkey Prefrontal Cortex Correlate with Aging-Related Cognitive Impairment. J Neurosci. 2010;30(22):7507-7515. doi:10.1523/JNEUROSCI.6410-09.2010
8 Xekardaki A, Kövari E, Gold G, et al. Neuropathological changes in aging brain. Adv Exp Med Biol. 2015;821:11-17. doi:10.1007/978-3-319-08939-3_6
9 Kovacs GG, Lee VM, Trojanowski JQ. Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol Zurich Switz. 2017;27(5):675-690. doi:10.1111/bpa.12536
10 Kovacs GG, Milenkovic I, Wöhrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol (Berl). 2013;126(3):365-384. doi:10.1007/s00401-013-1157-y
11 Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837-1844. doi:10.1212/01.wnl.0000219668.47116.e6
12 attson MP, Arumugam TV. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018;27(6):1176-1199. doi:10.1016/j.cmet.2018.05.011
13 Browder KC, Reddy P, Yamamoto M, et al. In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat Aging. Published online March 7, 2022:1-11. doi:10.1038/s43587-022-00183-2
14 Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-676. doi:10.1016/j.cell.2006.07.024
15 Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861-872. doi:10.1016/j.cell.2007.11.019
16 Miura Y, Li MY, Revah O, Yoon SJ, Narazaki G, Pașca SP. Engineering brain assembloids to interrogate human neural circuits. Nat Protoc. 2022;17(1):15-35. doi:10.1038/s41596-021-00632-z
17 Kelley KW, Pașca SP. Human brain organogenesis: Toward a cellular understanding of development and disease. Cell. Published online November 12, 2021:S0092-8674(21)01177-6. doi:10.1016/j.cell.2021.10.003
18 Collins FS, Varmus H. A New Initiative on Precision Medicine. N Engl J Med. 2015;372(9):793-795. doi:10.1056/NEJMp1500523
19 A G, Sj Y, Ss T, et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat Neurosci. 2021;24(3). doi:10.1038/s41593-021-00802-y
20 Jezierski A, Baumann E, Aylsworth A, et al. Electrophysiological- and Neuropharmacological-Based Benchmarking of Human Induced Pluripotent Stem Cell-Derived and Primary Rodent Neurons. Stem Cell Rev Rep. 2022;18(1):259-277. doi:10.1007/s12015-021-10263-2
21 Nimtz L, Hartmann J, Tigges J, et al. Characterization and application of electrically active neuronal networks established from human induced pluripotent stem cell-derived neural progenitor cells for neurotoxicity evaluation. Stem Cell Res. 2020;45:101761. doi:10.1016/j.scr.2020.101761
22 Sabate-Soler S, Nickels SL, Saraiva C, et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia. Published online March 9, 2022. doi:10.1002/glia.24167
23 Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115-130. doi:10.1038/nrd.2016.245
24 Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322-333. doi:10.1056/NEJMoa1304839
25 Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341-350. doi:10.1056/NEJMoa1210951
26 Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311-321. doi:10.1056/NEJMoa1312889
27 Engle SJ, Blaha L, Kleiman RJ. Best Practices for Translational Disease Modeling Using Human iPSC-Derived Neurons. Neuron. 2018;100(4):783-797. doi:10.1016/j.neuron.2018.10.033
28 Vincent F, Loria P, Pregel M, et al. Developing predictive assays: the phenotypic screening “rule of 3.” Sci Transl Med. 2015;7(293):293ps15. doi:10.1126/scitranslmed.aab1201
29 Wehling M. Assessing the translatability of drug projects: what needs to be scored to predict success? Nat Rev Drug Discov. 2009;8(7):541-546. doi:10.1038/nrd2898
30 Hardingham GE, Pruunsild P, Greenberg ME, Bading H. Lineage divergence of activity-driven transcription and evolution of cognitive ability. Nat Rev Neurosci. 2018;19(1):9-15. doi:10.1038/nrn.2017.138
31 Hawrylycz M, Miller JA, Menon V, et al. Canonical genetic signatures of the adult human brain. Nat Neurosci. 2015;18(12):1832-1844. doi:10.1038/nn.4171
32 Sloan SA, Darmanis S, Huber N, et al. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron. 2017;95(4):779-790.e6. doi:10.1016/j.neuron.2017.07.035
33 Bardy C, van den Hurk M, Kakaradov B, et al. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol Psychiatry. 2016;21(11):1573-1588. doi:10.1038/mp.2016.158
34 Dolmetsch R, Geschwind DH. The human brain in a dish: The promise of iPSC-derived neurons. Cell. 2011;145(6):831-834. doi:10.1016/j.cell.2011.05.034
35 Ryan SK, Gonzalez MV, Garifallou JP, et al. Neuroinflammation and EIF2 Signaling Persist despite Antiretroviral Treatment in an hiPSC Tri-culture Model of HIV Infection. Stem Cell Rep. 2020;14(5):991. doi:10.1016/j.stemcr.2020.04.006
36 De Sousa PA, Steeg R, Wachter E, et al. Rapid establishment of the European Bank for induced Pluripotent Stem Cells (EBiSC) - the Hot Start experience. Stem Cell Res. 2017;20:105-114. doi:10.1016/j.scr.2017.03.002
37 Li M, Zhao H, Ananiev GE, et al. Establishment of Reporter Lines for Detecting Fragile X Mental Retardation (FMR1) Gene Reactivation in Human Neural Cells. Stem Cells Dayt Ohio. 2017;35(1):158-169. doi:10.1002/stem.2463
38 Roberts B, Haupt A, Tucker A, et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol Biol Cell. 2017;28(21):2854-2874. doi:10.1091/mbc.E17-03-0209
39 Zhong S, Zhang S, Fan X, et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature. 2018;555(7697):524-528. doi:10.1038/nature25980
40 Raj B, Wagner DE, McKenna A, et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat Biotechnol. 2018;36(5):442-450. doi:10.1038/nbt.4103
41 Doss MX, Sachinidis A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells. 2019;8(5):403. doi:10.3390/cells8050403
42 Shibuya Y, Kumar KK, Mader MMD, et al. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci Transl Med. 2022;14(636):eabl9945. doi:10.1126/scitranslmed.abl9945
43 Kooreman NG, Kim Y, de Almeida PE, et al. Autologous iPSC-based Vaccines Elicit Anti-Tumor Responses in Vivo. Cell Stem Cell. 2018;22(4):501-513.e7. doi:10.1016/j.stem.2018.01.016
44 Cai J, Li W, Su H, et al. Generation of Human Induced Pluripotent Stem Cells from Umbilical Cord Matrix and Amniotic Membrane Mesenchymal Cells*. J Biol Chem. 2010;285(15):11227-11234. doi:10.1074/jbc.M109.086389
45 NIH. Genotype-Tissue Expression Program (GTEx). https://commonfund.nih.gov/GTex